Of Go, C, Perl and fastq file conversion Vol IV : gone in 60 seconds (or less)
In the final part of this series, we will test the performance of the four parsers, in a scenario emulating the batch analysis of sequencing data. We will use the sample fastq file 3_OHara_S2_rbcLa_2019_minq7.fastq from https://zenodo.org/record/3736457. This is a 35MB file of 21791 long sequences for a nanopore experiment. Download the data and save them to a directory in your hard disk. Then use the following bash time_fastq2a_shell.txt (change the extension to .sh before running!) to process this file 500 times with each of the four methods : seqtk (C), seqkit (Go), perl - regex (code presented here saved into fastq2a_regex_comp_in_func.pl file) and perl - flag (code presented there saved into the fastq2a_flag.pl file).
library Mean SD
seqtk 0.08651800 0.05804526
seqkit 0.10549900 0.02889980
flag 0.09279242 0.01132046
regex 0.12050400 0.01370685and a boxplot is used to show the variability of individual runs: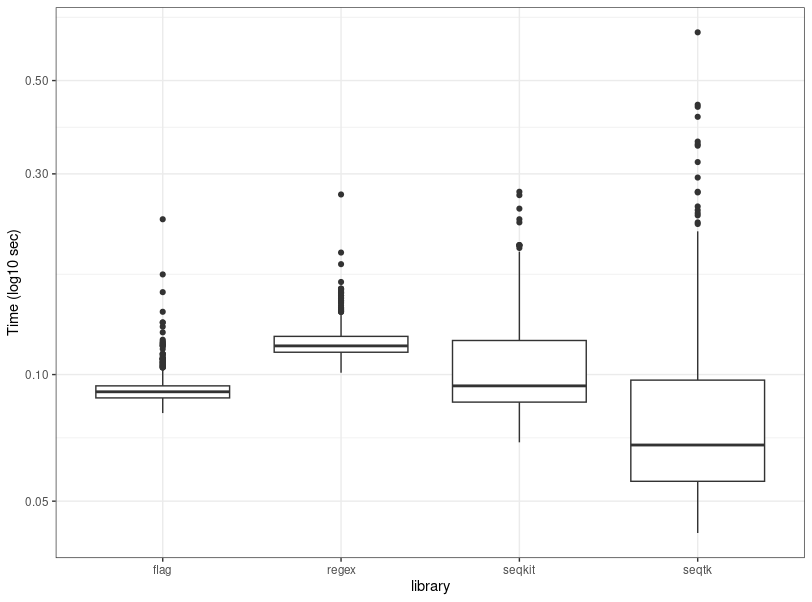
It should not be surprising that the compiled C library achieves the best (but variable) performance, followed closely by the flag based perl version (slower by 7%), the go library (slower by 22% relative to C) and finally the regex perl version. The variability of the Perl parsers was between 2.5 to ~5 less than those of the compiled languages. One of the benefits of the Perl parsers is that they don't have to output to a file, but can spit the results in the namespaces of other perl applications that perhaps use BioPerl, reducing thus the downstream IO overhead for moving results around.
 I like to use Perl for material other than text.
I like to use Perl for material other than text.
It's amazing how well perl performs when avoiding Regex