Extracting coronavirus SARS-CoV-2 spike protein sequences with BioPerl
As I started learning Perl I found out that there is a collection of Perl modules for bioinformatics tasks and it is called BioPerl. Intrigued I decided to try to use it for a simple task as it would help me in learning Perl. So the next question was: what task should I choose? Maybe download file with biological sequences, parse it and filter according some criteria? Thinking about this further I made a choice. I will work with sequences of infamous coronavirus which is more precisely named as SARS-CoV-2!
Spikes of coronaviruses – why are they so important?
If you look at pictures of coronaviruses you will see that they resemble the crowns of the royal family members that were magically shrunken 2,000,000 times. The spikes on the outside of the virus are made of a protein that is called “Spike glycoprotein”. This “S protein” or “spike protein” binds to the receptors on the membranes of our cells and is necessary for virus to get inside the cell. “To get in, or not to get in“: that is the question as viruses can replicate only by infecting the cells. One can imagine that the coronavirus without the spikes is a miniscule harmless jewelry floating around.
Let’s download the data and take a look at it
For the task I used Perl v5.30 PDL edition on Windows and installed all the necessary modules with cpanm. I downloaded the fasta file with coronavirus protein sequences by using NCBI Search and saved it as “corona_protein_seqs.fasta”. To begin with I needed to load all the modules that will be needed:
use v5.12;
use Bio::SeqIO;
use Chart::GGPlot qw(:all);
use Alt::Data::Frame::ButMore;
use Data::Frame;
use PDL;
Next I went on to read the input fasta file and create a new fasta file for output:
my $seqio_in = Bio::SeqIO->new(-file => "corona_protein_seqs.fasta",
-format => "fasta" );
my $seqio_out = Bio::SeqIO->new(-file => ">corona_spike.fasta",
-format => "fasta" );
Then the time came to think about how to filter spike proteins from all the proteins that virus could have. I kept in mind that the names of the sequences and their descriptions are stored in lines that start with a symbol “>”. Let’s look at several of these lines:
$ perl –ne "print if /^>/" corona_protein_seqs.fasta | head –n 5
>QIA20052 |nucleocapsid phosphoprotein [Severe acute respiratory syndrome coronavirus 2]
>QIA20048 |ORF6 protein [Severe acute respiratory syndrome coronavirus 2]
>QIA20053 |ORF10 protein [Severe acute respiratory syndrome coronavirus 2]
>QIA20049 |ORF7a protein [Severe acute respiratory syndrome coronavirus 2]
>QIA20051 |ORF8 protein [Severe acute respiratory syndrome coronavirus 2]
From this it is seen that protein description is after symbol "|" and name of a virus is inside square brackets.
Let’s find all the SARS-CoV-2 spike protein sequences
Next, I wanted to make sure that the sequences belong to the virus "Severe acute respiratory syndrome coronavirus 2" so proceeded like this:
while ( my $seq = $seqio_in->next_seq ) {
# Extracting description of a protein}
$id = $seq->desc;
# Selecting only the sequences that belong to SARS-CoV-2
if ($id =~ /\[Severe acute respiratory syndrome coronavirus 2\]/){
## }
Well, all of this helped to select the protein sequences that belong to coronavirus. But how to select only spike (S) glycoprotein? I made use of regexes that involved words "S", "spike" or "glycoprotein":
# Selecting only spike protein sequences
if ( $id =~ m/^(.*S[^\w].*)$/ or $id =~ m/spike|glycoprotein/i ) {
##
}
Let’s identify and remove partial sequences that are shorter
Not all the sequences that are saved in a NCBI database span all the protein. Luckily, with BioPerl it is easy to calculate lengths of the sequences and save them to separate array like "@seq_lengths":
# Selecting only spike protein sequences
if ( $id =~ m/^(.*S[^\w].*)$/ or $id =~ m/spike|glycoprotein/i ) {
# Pushing lengths of the sequences to an array
push @seq_lengths, $seq->length;
}
For visualization of lengths of the sequences I decided to create a histogram plot. I chose Chart::GGPlot module which is based on Hadley Wickham's ggplot2 in R. It went like this:
# Creating dataframe that will be used for plotting:
my $df = Data::Frame->new( columns => [
x => pdl(\@seq_lengths) ,
] );# Creating histogram plot:
my $p = ggplot( data => $df )
->geom_histogram( mapping => aes( x => 'x' ) )
->labs(
title => 'Histogram of spike protein sequences by length',
x => 'Sequence length',
y => 'Counts',
);
$p->show();
Plot was opened in the browser and looked like this:
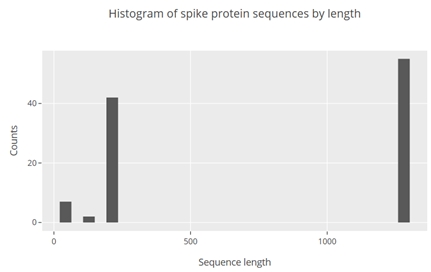
From the plot it is seen that the cutoff of 1000 could be applied to remove partial sequences. Knowing this it becomes easy to select the full-length spike protein sequences that could be used for further analysis.
 I blog about Perl.
I blog about Perl.{kind=link}
Leave a comment